
NEWS Persons with the serious disorder ALS, can have a genetic mutation that causes the protein SOD1 to aggregate in motor neurons in the brain and spinal cord. Researchers at Umeå University have discovered that, when injected into mice, the SOD1 aggregation spreads rapidly leading to ALS. The discovery has been described in the Journal of Clinical Investigation.
ALS, Amyotrophic lateral sclerosis, is a disorder which causes death of motor neurons in the brain and the spinal cord in charge of controlling muscles. This leads to progressive paralysis and death, often due to carbon dioxide narcosis.
“The occurrence of SOD1 aggregates in nerve cells in ALS patients has been known for a while,” says Thomas Brännström, professor of Pathology at Umeå University and one of the authors of the article. “But it has long been unclear what role the SOD1 aggregates play in the disease progression in humans carrying hereditary traits for ALS. We have now been able to show that the SOD1 aggregates start a domino effect that rapidly spreads the disease up through the spinal cord of mice. We suspect that this could be the case for humans as well.”

Researchers at the Departments of Medical Biosciences and Pharmacology and Clinical Neuroscience at Umeå University have examined whether the accumulation of the SOD1 aggregates inside nerve cells drives the ALS disease or if it is a harmless side-effect. The research team has been able to identify two different kinds of SOD1 aggregates in mice. The results showed that both kinds caused a spread of SOD1 aggregation when a small amount was injected into the spinal cord of mice. The accumulation of aggregates progressed in nerve cells along the entire spinal cord of the mice at the same time as it led to a rapid and eventually mortal outcome of the disease.

“The results show that the aggregation of SOD1 plays a critical role in the disease progression – a research hypothesis that ALS researchers in Umeå long has based its work upon,” says Stefan Marklund, professor of Clinical Chemistry. “More research is necessary, but our aim is to develop interventions that prevent or stop the fatal course of the disease in carriers of hereditary traits of ALS.”
Read a digital publication of the article in the Journal of Clinical Investigation
The Journal of Clinical Investigation, article: Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. Authors: Elaheh Ekhtiari Bidhendi, Johan Bergh, Per Zetterström, Peter M. Andersen, Stefan Marklund and Thomas Brännström. DOI: 10.1172/JCI84360.
Thomas Brännström, Department of Medical Biosciences, Umeå UniversityPhone: +46 90-785 15 23; +46 70-634 39 32
Email: thomas.brannstrom@umu.se
Portrait photo for download
Stefan Marklund, Department of Medical Biosciences, Umeå UniversityPhone: +46 90-785 12 39; +46 70-568 77 03
Email: stefan.marklund@umu.se
Portrait photo for download
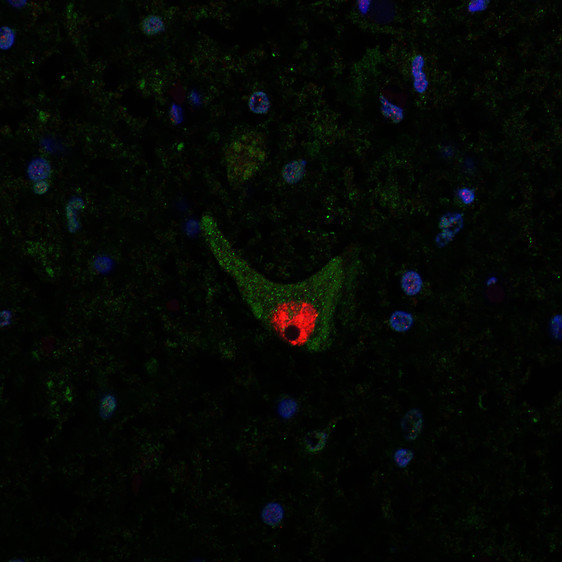
Caption: Aggregated SOD1 protein can be seen as green dots in the cell fluid of a motor neuron in the spinal cord of an ALS patient. The red part is the nerve cell nucleus. Credit: Peter Andersen.
Editor: Anna Lawrence