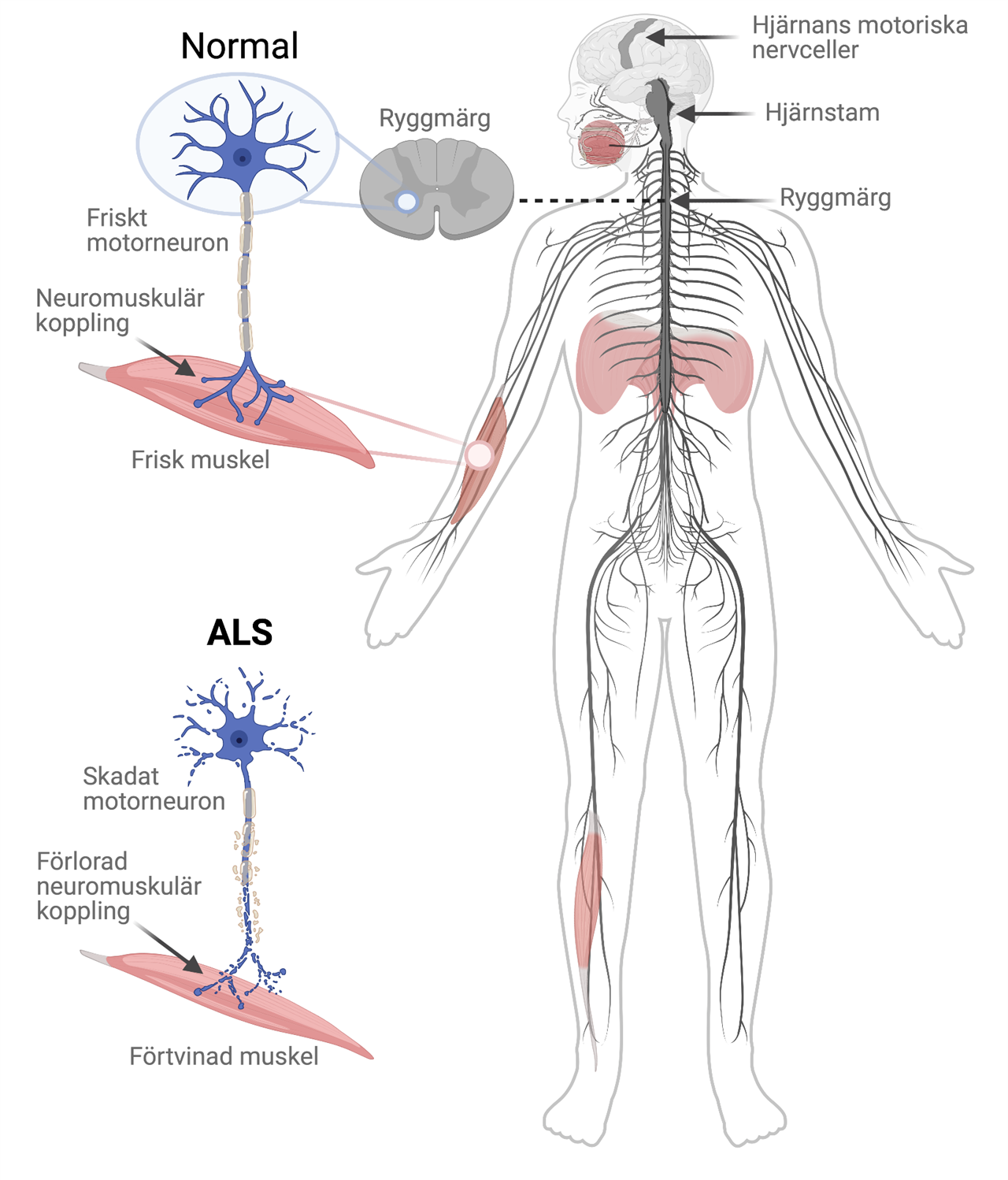
Amyotrofisk lateralskleros, ALS, är ett samlingsnamn på en grupp neurodegenerativa sjukdomar som leder till att motoriska nervceller i ryggmärgen och hjärnan dör.
ALS kännetecknas av en fortskridande förlamning som oftast börjar i en hand, en fot eller i ansiktet. Huvudproblemet vid ALS är att nervcellerna i nervsystemet som styr våra muskler bryts ned. Allt eftersom de motoriska nervcellerna slutar signalera till musklerna blir de allt svagare, förtvinar och blir så småningom förlamade.
BildIllustration skapad av Ulrika Nordström med BioRender.com
Både hjärnans och ryggmärgens motorneuroner kan drabbas. De övre motorneuronen sitter i hjärnbarken. När dessa celler dör får hjärnan svårt att koordinera och slappna av i muskler. Detta gör att den drabbade personen får svårt med finmotoriken.
De nedre motorneuron sitter i hjärnstammen och på framsidan av ryggmärgen, från nacken ner till svanken. Dessa nervceller har en direkt förbindelse med muskelfibrer i olika delar av kroppen, och får dessa att dra ihop sig när vi vill böja en arm, ett ben, ett finger, etc. När dessa nervceller dör så blir muskeln svag och förtvinar.
Hos ALS-patienter ser man nästan alltid symptom på förlust av både övre och nedre motorneuron. Patienterna är alltså både svaga med förtvinade muskler, och stela och svårkoordinerade i andra muskler.
I regel startar sjukdomen väldigt lokalt, i en hand eller en fot. Med tiden får patienten även besvär högre upp på armen/benet och symtomen sprider sig ”utifrån” och in mot bålen. Ett tag efter att sjukdomen startat får man ofta besvär från en annan extremitet på samma kroppshalva, om sjukdomen började i vänster hand kan man därefter få besvär med vänster fot. Därefter brukar sjukdomen spridas till den andra kroppshalvan. Ibland börjar sjukdomen i huvudets muskler, då får man svårt att artikulera, tugga och svälja mat och dryck.
Oavsett om sjukdomen börjar i ansiktet eller i en arm eller ben så sprids sjukdomen med tiden till nervcellerna som styr andningsmuskulaturen och när andningsfunktionen blivit mycket dålig avlider man i andningssvikt.
De flesta ALS-patienter har välbevarade tankeförmågor även i sjukdomens slutskede och är, frånsett sin förlamning, samma person som de alltid varit. Hos en del patienter händer det dock att tankeförmågan och känsloregleringen till viss del blir påverkad. Det kan yttra sig som diskreta förändringar i språket, som att man har svårt att formulera långa komplicerade meningar (detta behöver alltså inte vara ett resultat av att man lätt blir andfådd, utan att man har svårt att formulera sig).
Hos en del patienter kan man få svårt att reglera sina känslor på ett normalt sätt utan kan skratta eller gråta ohejdat i oväntade situationer. De här personlighetsförändringarna, när man är drabbad, kan göra samspelet mellan patient, närstående och assistenter komplicerad, eftersom det kan vara svårt att skilja på reaktioner inför det faktum att man har en obotlig, dödlig sjukdom, från reaktioner som beror på sjukdomens angrepp på hjärnan.
De flesta patienter som drabbas av ALS avlider inom tre till fyra år efter de första symtomen uppträder, en del patienter lever dock mycket längre än så. Det finns långtidsöverlevare, där sjukdomen av oförklarliga skäl utvecklas mycket långsamt och dessa patienter kan leva 10 till 20 år med sjukdomen.
De flesta som insjuknar i ALS är i 60-årsåldern och det är något vanligare att män drabbas. ALS är dock en mycket varierande sjukdom och ovanliga fall av ALS har rapporterats hos såväl spädbarn som hos mycket gamla (över 90 år) patienter. Man brukar säga att risken att någon gång i livet insjukna i ALS är ungefär 1 på 400 för en genomsnittlig person. Vi vet att rökning ökar risken något och att sjukdomen är lite vanligare hos jordbrukare och elektriker, men den individuella, personliga risken att insjukna om man tillhör ett riskyrke är ändå låg.
Ungefär 10–15 procent av alla ALS-patienter har en ärftlig form av ALS. De har en släkting som har haft ALS och sjukdomsanlaget nedärvs från en generation till nästa, så kallad familjär ALS (F-ALS). I sådana släkter kan man ofta identifiera en enskild mutation i en gen som orsakar sjukdomen och nedärvs tillsammans med den.
Genetiska studier har identifierat förändringar i ett fyrtiotal olika gener som kopplats samman med ALS. Mutationer i dessa gener har hittats både hos patienter med F-ALS och patienter med diagnosen S-ALS. Det finns genetiska förändringar som orsakar ALS. Andra kan öka eller minska risken att drabbas av ALS.
De två vanligast förekommande förändringarna som är förknippade med ALS finns i generna C9orf72 och SOD1.
De flesta personer som blir sjuka i ALS (85–90 procent) har inte kännedom om någon nära släkting som varit sjuk i ALS och dessa fall av sjukdomen brukar kallas sporadisk ALS (S-ALS).
Orsaken till majoriteten av alla ALS diagnoser är fortfarande okänd. De många olika ärftliga genetiska förändringar som kopplats till ALS och studier av den normala funktionen för de proteiner de kodar för, har dock identifierat en rad cellulära förändringar som kan bidra till sjukdomsutveckling.
Geners uttryck och många cellulära funktioner kan också förändras med stigande ålder och på grund av olika miljöfaktorer. Nedärvda genförändringar som kan orsaka ALS betyder ju inte att man föds med ALS. Sjukdomen utvecklas vanligtvis inte förrän i 40- till 60-årsåldern. Det krävs alltså mer än bara en genförändring som resulterar i felaktiga proteiner för att utveckla ALS. Det är först när cellen måste kompensera för ytterligare ålders-, eller miljöfaktorers påverkan som ansamlingar av felaktiga proteiner blir överväldigande och leder till sjukdomsutveckling.
Läs mer om vanliga genetiska orsaker och familjära former av ALS
Det finns ett läkemedel som används rutinmässigt vid ALS – Rilutek. Det är en tablett som tas om dagligen så fort man ställt diagnosen. Exakt hur Rilutek fungerar är inte känt idag, men man vet att behandlingen saktar ned sjukdomen en aning och att den ger patienten ett par månader längre att leva. Ju tidigare Rilutek sätts in desto bättre prognos och fördröjs sjukdomsförloppet. Det finns ett annat läkemedel vid namn Edaravone som är godkänt i vissa länder, däribland USA och Japan. Det verkar kunna sakta ned symtomutvecklingen en aning hos vissa specifika patientgrupper, men till skillnad från Rilutek ges Edaravone som i dropp direkt till blodbanan. Det pågår läkemedelsstudier i Europa som ska klargöra om det är lämpligt att godkänna Edaravone inom EU och om man kan förenkla rutinerna kring hur läkemedlet ska ges. I Sverige har Rådet för nya terapier (NT-rådet), en expertgrupp hos Sveriges Kommuner och regioner (SKR), fattat beslut om att inte införa Edaravone som behandlingsterapi för ALS-patienter fram till dess att det framkommit mer dokumentation om säkerhet, effekt och kostnadseffektivitet.
Läs mer om läkemedelsprövningar
Som ALS-patient blir man mer och mer beroende av sin omgivning för att klara av sin vardag. Tillsammans med assistenter, logopeder och arbetsterapeuter kan man dock få hjälp att hitta nya strategier som hjälper till att behålla patientens autonomi så långt det är möjligt. När sjukdomen på allvar börjat påverka förmågan till att äta och andas normalt kan man få hjälp att förstärka energi- och näringsintaget och andas mer effektivt med en så kallad icke-invasiv ventilation. Sådana insatser kan tyckas små, men har visat sig ge större överlevnadsvinster än något läkemedel kunnat leverera.
Sammanfattningsvis är ALS en sjukdom som bryter ner specifika delar av nervsystemet, vilket leder till förlamning, stort beroende av omgivningen och till slut döden.



