Den vanligaste kända orsaken till ALS och frontotemporal demens (FTD) är en förändring i C9orf72 genen. C9orf72 är alltså en gen som är kopplad till mer än en sjukdom. Som bärare av C9orf72-förändringar har man en kraftigt förhöjd risk (i princip 90%) att drabbas av antingen ALS eller frontallobsdemens (FTD). FTD är en variant av demens där minnesförmågan är bevarad tidigt i sjukdomen men där personens omdöme, empati och förmåga till långsiktig planering blir starkt påverkad. Ibland drabbas en patient av både ALS och FTD, t ex kan man i början ha enbart ALS-symtom och halvvägs genom sjukdomen drabbas man även av FTD.
C9orf72 genen identifierades 2011 och ledde till ett stort genombrott. Hela 14% av våra ALS patienter har denna mutation, och ungefär 4-5% av de FTD patienter som analyserats. Ett annat namn på just C9orf72HRE-ALS är Mörtsellsjukan, efter Johannes Mörtsell, präst och riksdagsman från Västerbotten, som avled 1888 med diagnosen progressiv spinal muskelatrofi. Han är det äldsta kända fallet av ALS i Norden. Släkten Mörtsell är den största studerade ALS/FTD-släkten i världen. Idag vet vi att C9orf72HRE nedärvs med sjukdomen och orsakar Mörtsellsjukan.
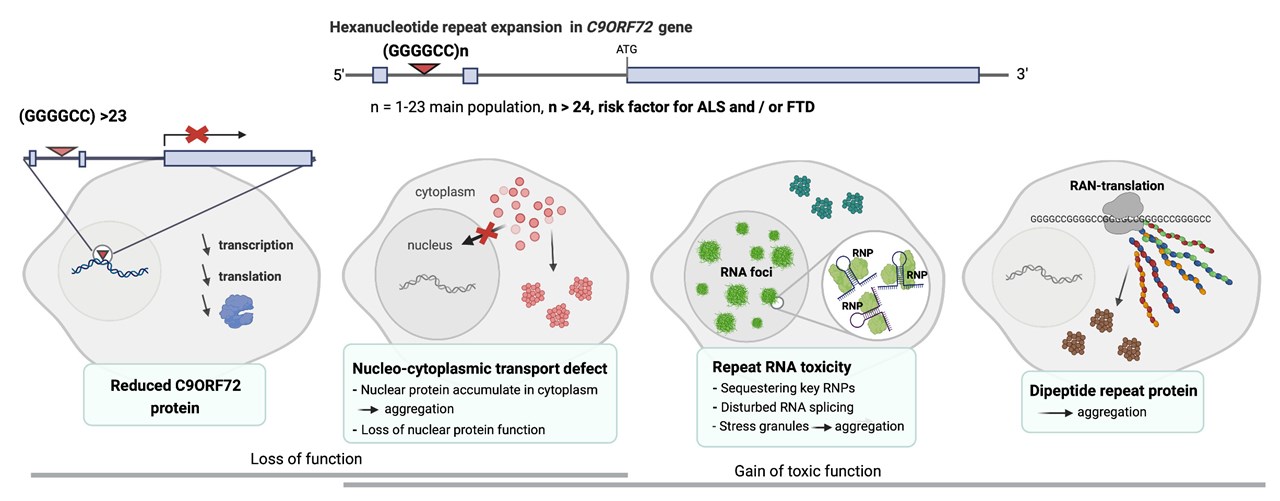
Genen C9orf72 kodar för tillverkning av proteinet med samma namn, C9ORF72. Proteinets normala funktion är ännu oklar men inkluderar reglering av transport av olika molekyler till olika delar i cellen (vesikeltransport). Den genförändring som kan orsaka ALS och FTD är en upprepning av en sekvens på sex nukleotider GGGGCC (hexanukleotid repetitiv expansion, HRE) i en region som reglerar uttryck av C9orf72-genen. Normalt har människor 1 till 23 upprepningar av GGGGCC-sekvensen, men hos 10-14 procent av de som har ALS finns flera hundra, ibland tusen, upprepningar.
C9orf72HRE-expansionen nedärvs dominant. Det betyder att det räcker med att en av genkopiorna på kromosom 9 har en expansion för att kunna orsaka ALS. Patienterna är därför bärare av både en expanderad C9orf72 sjukdomsgen och en normal C9orf72 gen. Anmärkningsvärt är att C9orf72HRE sjukdomsgenen har olika genomslag i olika släkter. Det är alltså inte alla som är bärare av genen som blir sjuka men bärare har kraftigt ökad risk att drabbas av ALS eller FTD. I en del familjer kan det vara vanligare att drabbas av FTD än ALS, men även sannolikheten för en bärare av genförändringen att drabbas av sjukdom kan variera mellan olika släkter.
Repetitiva DNA sekvenser gör genen instabil och när DNA:t skall kopieras inför celldelning händer det ofta att dottercellen får ännu fler upprepningar. Antalet upprepningar kan därför variera i olika organ i en människa och våra studier har visat att de delar av nervsystemet som drabbas vid ALS ofta har längre repetitiva sekvenser än till exempel blodceller. Det är möjligt att även personer som har lågt, normalt, antal upprepningar av GGGGCC-sekvensen i blodceller (som normalt används för genetiska analyser på patienter) faktiskt har en betydligt längre sekvens i delar av det centrala nervsystemet. Mer forskning krävs för att ta reda på hur vanligt förkommande det är med expansion av den upprepade sekvensen i nervceller och om C9orf72 möjligen kan orsaka ännu fler fall av ALS än vi kan upptäcka genom blodanalyser.
BildIllustration skapad av Ulrika Nordström med BioRender.com
Mutationer i SOD1-genen är den näst vanligaste kända orsaken till ALS. Det var den första orsakande faktor som associerades med ALS. Studier av familjer med en ärftlig variant av ALS (F-ALS) gjorde att man 1993 fann två olika mutationer i SOD1 genen som visades gå i arv tillsammans med ALS sjukdom. SOD1 mutationer orsakar cirka 20% av alla ärftliga fall av ALS, men kan också förekomma i S-ALS patienter (de som inte har kännedom om tidigare fall inom familjen). SOD1-mutationer identifieras i cirka 2% av alla ALS patienter.
Idag har forskare identifierat mer än 200 olika mutationer i SOD1 genen hos ALS patienter. Det behöver inte betyda att alla dessa mutationer verkligen orsakar ALS, men det är troligt att de flesta bidrar till, och ökar risken för sjukdomsutveckling.
Olika mutationer i SOD1 genen kan ge upphov till olika sjukdomsbild och överlevnadstider. Vissa mutationer i SOD1 associeras till en aggressiv sjukdomsutveckling med en medelöverlevnadstid på 1,5 år efter diagnos. Andra specifika mutationer associeras till en exceptionellt långsam försämring med överlevnadstid som ofta överskrider 10 år.
De flesta SOD1 mutationer som kopplats till ALS nedärvs dominant. Det betyder att det räcker med att en av SOD1 genkopiorna på kromosom 21 har mutationen för att den ska orsaka eller öka risken för att utveckla ALS.
Genomslagskraften för olika mutationer varierar, dvs. om förändringen leder till sjukdom eller ej. Genetiska förändringar med hög genomslagskraft orsakar sjukdom hos de flesta bärare, medan låg genomslagskraft bara ökar risken att drabbas i olika grad. Sjukdomsutveckling kan därför hoppa över en eller två generationer. Genomslagskraften av en och samma mutation kan även variera mellan olika släkter. Genom att studera skillnader mellan olika släkter med varierande genomslagskraft hoppas vi kunna identifiera ytterligare faktorer som bidrar till, eller skyddar mot sjukdomsutveckling.
Den vanligast förekommande SOD1 mutationen kallas D90A och identifierades 1993 av Peter Andersen och hans kollegor här vid Umeå Universitet. Just denna mutation är särskilt vanlig i Sverige och är bäst studerad i den norrländska populationen där den bara orsakar ALS om man har mutationen i båda kopiorna av SOD1-genen.
SOD1 genen kodar för proteinet Superoxiddismutas 1 (SOD1). SOD1 proteinet finns i alla celler och utgör 1-2% av det totala lösliga proteinet i centrala nervsystemet. När ett nytt SOD1 protein tillverkas ska det modifieras och packats ihop till dess specifika 3-dimensionella struktur som är essentiell för dess normala funktion, vilket är att neutralisera fria radikaler (superoxider) som är extremt skadliga för cellen.
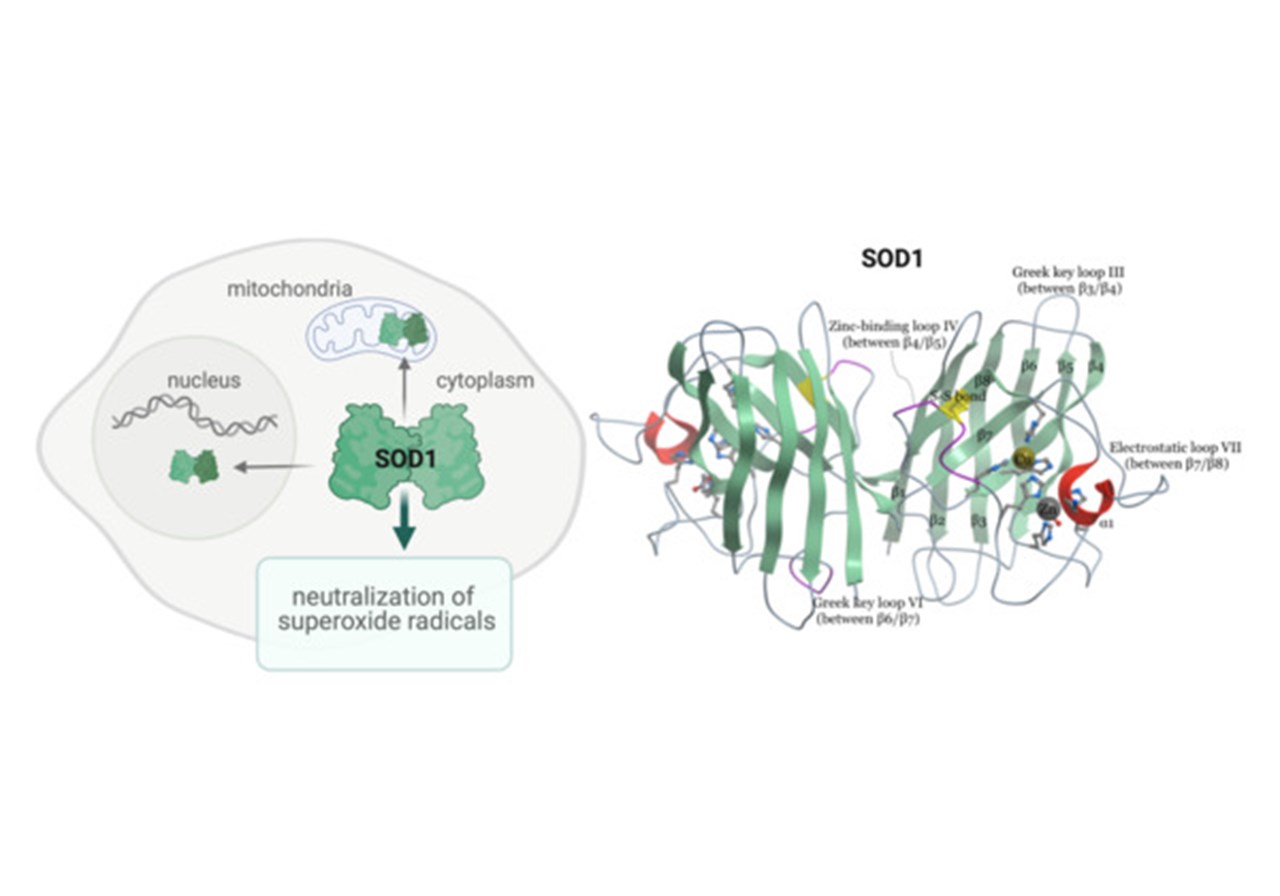
SOD1 proteinets normala struktur och funktion SOD1 proteinet är huvudsakligen lokaliserat till cellens cytoplasma, men finns även i mitokondrier (cellens energifabriker) och cellkärnan som innehåller arvsmassan (DNA). Normalt SOD1 protein modifieras och formas till en extremt stabil, 3-dimensonell struktur. Sedan binder två SOD1 proteiner varandra och bildar det enzym som neutraliserar skadliga superoxidjoner.
BildIllustration skapad av Ulrika Nordström med BioRender.comStudier har visat att mutationer i SOD1 bidrar till ALS genom att proteinet får en ny toxisk funktion snarare än att det beror på minskad normal SOD1 funktion. Denna toxiska ”gain-of-function” anses bero på att muterat SOD1 protein lättare tappar sin normala form, felveckas och klistras ihop i felaktiga former.
I studier på vävnad från avlidna, obducerade patienter kan man med hjälp av antikroppar, som binder felaktigt veckat, SOD1-protein, påvisa ansamlingar av SOD1-protein i cytoplasman i motoriska nervceller och de omkringliggande stödjecellerna (sk gliaceller). Ansamlingar av felvecklat SOD1-protein är vanliga i det motoriska nervsystemet hos ALS-patienter som är bärare av en mutation i SOD1, men kan även ses hos patienter med sporadisk ALS.
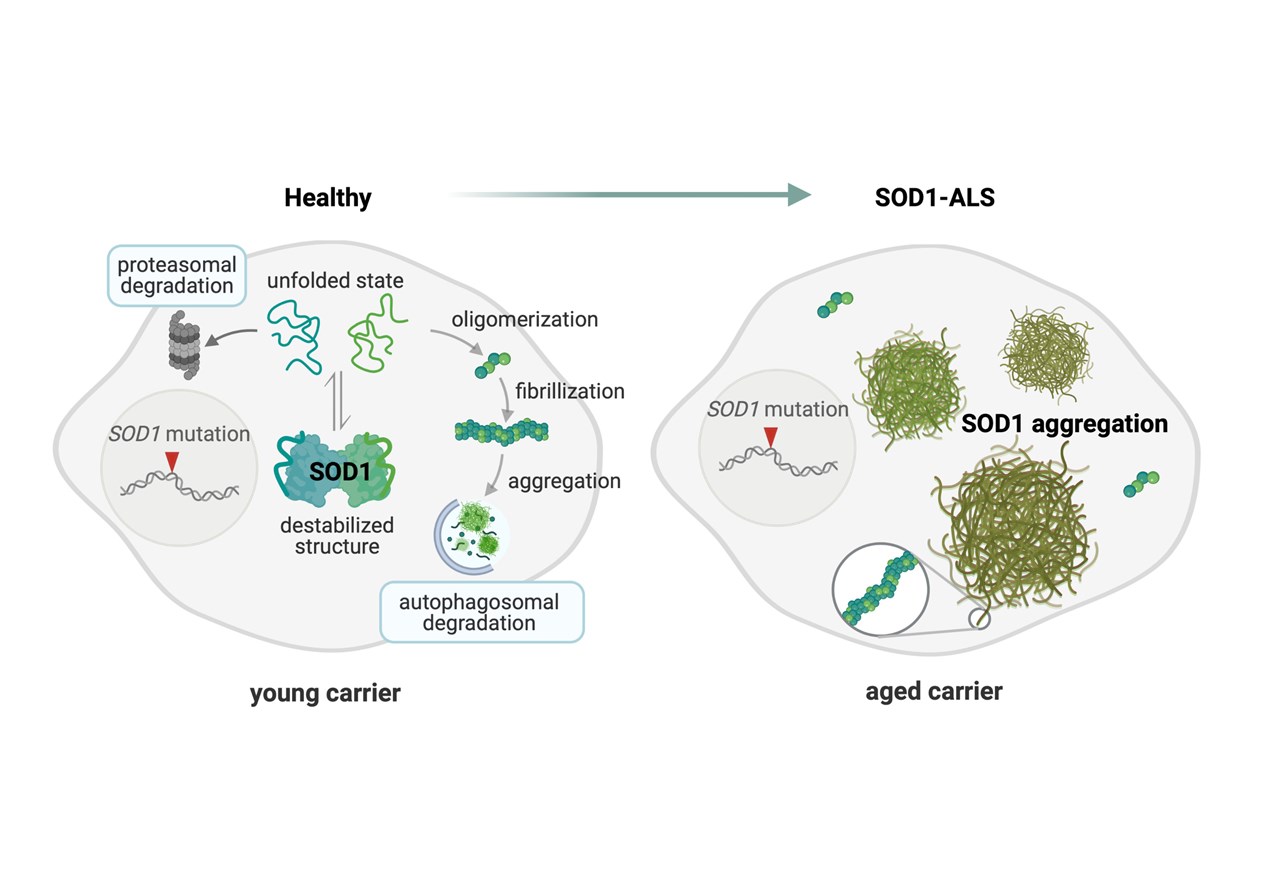
Vad händer i cellerna vid SOD1-ALS? Många mutationer som kan orsaka ALS försvagar stabiliteten eller förhindrar korrekt proteinstruktur. Ostrukturerade SOD1 proteiner och börjar lätt klistra ihop sig med varandra och bildar skadliga ansamlingar av felaktigt format SOD1.
BildIllustrationen är skapad av Illustration skapad av Ulrika Nordström med BioRender.comVad som orsakar felveckning av SOD1 hos patienter med sporadisk ALS eller hos patienter med mutation i andra gener än SOD1 är i dag okänt. En riskfaktor skulle kunna vara nedsatt cirkulation och syresättning i nervsystemet. Varje SOD1 protein innehåller en disulfidbindning (mellan aminosyrorna cystein 57 och cystein 146). Disulfidbindningen ökar SOD1-proteinets stabilitet. Vår forskning på celler från ALS patienter har visat att låg syrehalt i celler skulle kunna resultera i förlust av disulfidbindning, minskad proteinstabilitet och ökad mängd skadliga ansamlingar av felstrukturerat SOD1.
Klumpar av SOD1 protein (färgat med brunt) i nervcell som sköter den motoriska funktionen i kroppen. Bilden kommer från en patient med ALS som saknar mutation i SOD1. Detta tyder på att proteinet är inblandat i sjukdomsmekanismen vid ALS även om mutation saknas i proteinet.
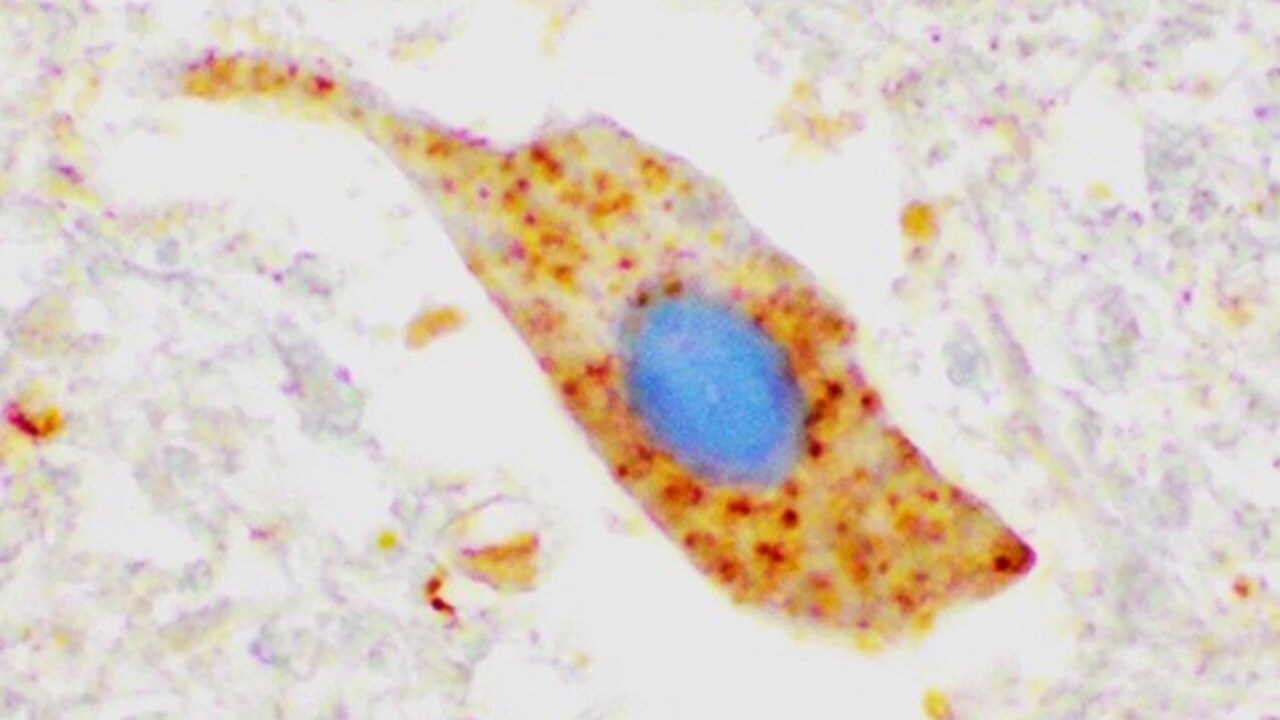
Klumpar av SOD1 protein (färgat med brunt) i nervcell som sköter den motoriska funktionen i kroppen. Bilden kommer från en patient med ALS som saknar mutation i SOD1. Detta tyder på att proteinet är inblandat i sjukdomsmekanismen vid ALS även om mutation saknas i proteinet.
BildKarin ForsbergKraftig fysisk aktivitet har diskuterats som riskfaktor för ALS men vissa studier har föreslagit ett samband medan andra inte kunde visa att extremidrott var en riskfaktor.
En annan bidragande orsak kan vara att cellens förmåga att städa bort felaktiga proteiner minskar med ökande ålder. Våra studier på celler från patienter visar att nedsatt nedbrytning av proteiner leder till ökad mängd ostrukturerat SOD1 som skulle kunna bidra till skadliga ansamlingar.
För att kunna studera vad som händer när SOD1 felveckas använder vi oss både av hudceller från patienter och transgena möss som sjukdomsmodeller. Transgent uttryck av en muterad variant som orsakar ALS i människor, men också kraftigt överuttryck av den normala SOD1-genen, resulterar i en ALS-liknande sjukdom och ansamlingar av felaktigt strukturerat SOD1 protein i det motoriska nervsystemet hos mössen.
Vi har identifierat två strukturellt olika typer av felaktigt format SOD1 som kan bildas och ansamlas i de ALS-sjuka mössens nervsystem. För att studera hur skadliga dessa är och hur de påverkar omgivande celler har vi tagit preparationer av SOD1 från sjuka möss och sprutat in i ryggmärgen på möss som bär på sjukdomsgenen men fortfarande är unga och friska.
Försöken visade att båda varianterna av felaktigt format SOD1 protein startade utveckling av ALS-sjukdom, som börjar med muskelförsvagning i det ben som får sina nervsignaler från området som injicerades. Symtomen spred sig senare i tur och ordning till musens övriga ben på ett sätt som liknar hur symtomen sprider sig hos ALS patienter. Samtidigt började det bildas nya ansamlingar av felaktigt formade SOD1 proteiner som hade precis samma struktur som den som sprutats in. Injektion av muterat SOD1-protein i preparationer av vävnad från obducerade ALS-patienter ger liknande resultat.
SOD1-protein kan anta en sjukdomsframkallande tredimensionell form som kan sprida sig vidare till nya celler i andra delar av nervsystemet och på så sätt orsaka motsvarande spridning av ALS symtom till olika kroppsdelar. De två strukturellt olika former av SOD1 som kan bildas i ALS-sjuka möss ger båda upphov till ALS-liknande sjukdom, men med olikheter i symtomutveckling och överlevnadstid. Skillnader mellan olika struktur på SOD1-aggregat hos olika patienter, skulle möjligen kunna bidra till skillnader i symtom, sjukdomsutveckling och överlevnad.



