År 1993 gjordes upptäckten att mutationer i genen som kodar för enzymet superoxiddismutas 1 (SOD1) kan orsaka amyotrofisk lateralskleros (ALS), en neurodegenerativ sjukdom som förlamar alla kroppens muskler. Samma år grundades ett forskningssamarbete med fokus på ALS vid Umeå Universitet och Norrlands universitetssjukhus (NUS) mellan Stefan Marklund från klinisk kemi, Thomas Brännström från klinisk patologi och Peter Andersen från klinisk neurologi. Målet var att studera SOD1 i relation till ALS för att förstå hur sjukdomen uppstår.
Sedan dess har samarbetet vuxit och ALS-forskningen har utvecklats till ett nätverk av forskningsgrupper vid Umeå Universitet som delar expertis och infrastruktur för att göra nya upptäckter om ALS. Vårt mål är att bidra med kunskap som kan bereda vägen för framtida behandlingar av sjukdomen.
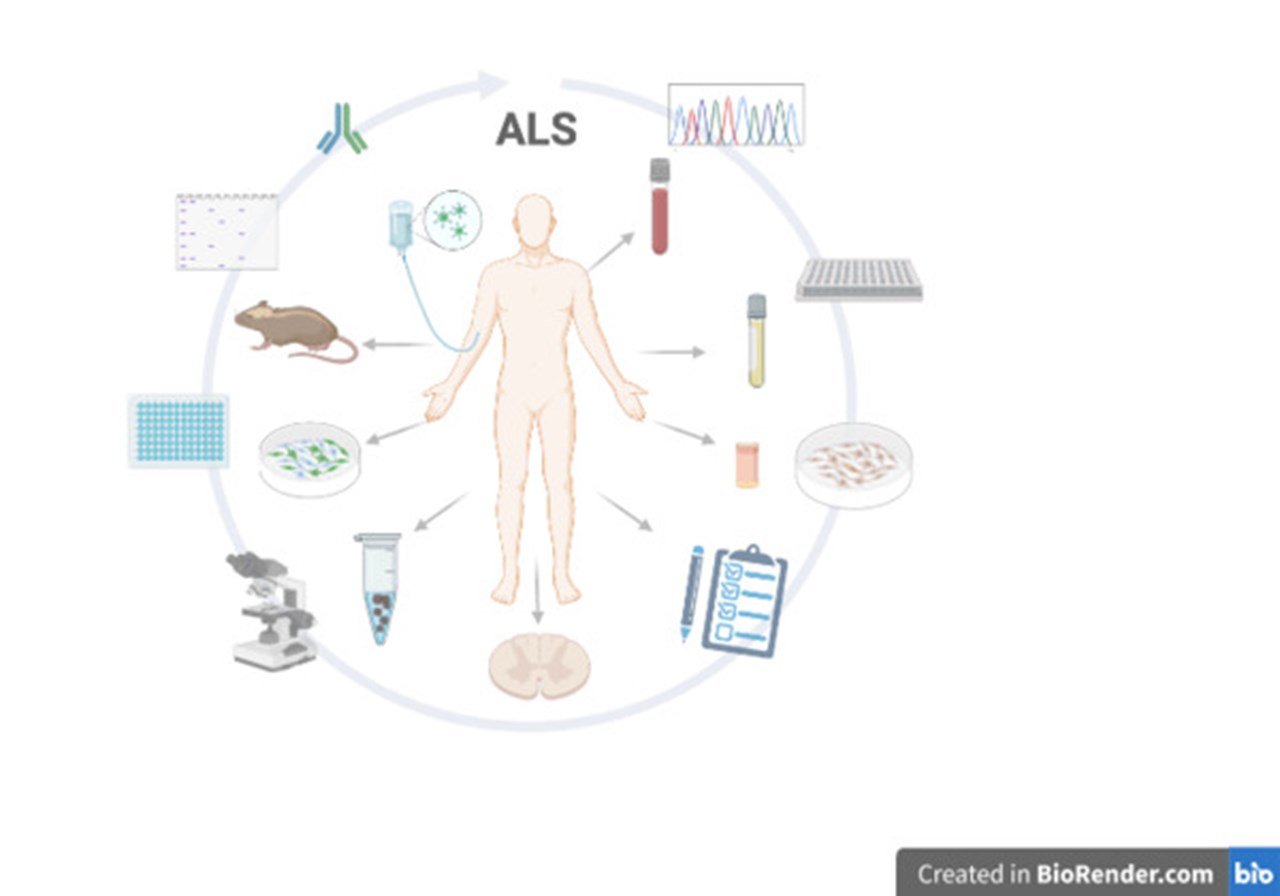
BildIllustration skapad av Ulrika Nordström med BioRender.com
Idag har vi byggt upp Sveriges största biobank med prover för ALS-forskning och är ledande svenskt centrum för ett flertal kliniska prövningar. Vi bedriver forskningsprojekt inom biokemi, genetik, histopatologi, cellbiologi, anatomi och klinisk neurologi, inklusive läkemedelsstudier med ALS-patienter.
Tillsammans har vi publicerat över 200 ALS-relaterade studier och består av cirka 25 medarbetare som brinner för ALS-forskning. I gruppen ingår laboratorieassistenter, doktorander, post-docs, forskningsingenjörer, forskningssjuksköterskor, läkare och professorer. De forskare som ingår i gruppen kommer från flera olika specialiteter däribland neurologi, kemi, patologi, molekylärbiologi och genetik.

Forskningssköterskan Lena Bylund, doktoranden och läkaren i neurofysiologi Ivar Winroth, forskningssköterskan Nina Sundqvist, professorn och överläkaren i neurologi Peter Andersen, forskningssköterskan Elisabeth Müller Granberg, forskningssköterskan Tina Björn och forskaren och specialistläkaren i neurologi Karin Forsberg utgör teamet på Neurologens forskningsavdelning på Norrlands universitetssjukhus.
Bild Mattias Pettersson
Forskningssjuksköterskan och studiekoordinatorn Lena Bylund tar hand om forskningsprover som har anlänt för analys.
Bild Mattias Pettersson
Forskningssjuksköterskan och studiekoordinatorn Tina Björn vid sin dator på kliniken.
Bild Mattias Pettersson
Forskningssjuksköterskan och studiekoordinatorn Nina Sundqvist i samspråk med läkaren och doktoranden Ivar Winroth på sitt arbetsrum.
Bild Mattias Pettersson
Forskningssjuksköterskan och studiekoordinatorn Elisabeth Müller Granberg i färd med att ta ett blodprov.
Bild Mattias Pettersson
Blodprover tas för användning såväl i den egna forskningen som i kliniska studier i samarbete med läkemedelsbolag.
Bild Mattias Pettersson
Forskningssjuksköterskan och studiekoordinatorn Nina Sundqvist mäter lungfunktionskapaciteten med hjälp av en spirometer.
Bild Mattias Pettersson
Ivar Winroth, läkare på Neurofysiologen och doktorand vid institutionen för klinisk vetenskap, enheten för neurovetenskaper, genomför det standardiserade kognitiva testet ECAS som många får gå igenom i samband med en ALS-utredning.
Bild Mattias Pettersson
Neurologen och forskaren Karin Forsberg och överläkaren i neurologi och professorn Peter Andersen diskuterar provsvar från en ALS-patient.
Bild Mattias PetterssonSom universitetssjukhus i Norra Regionen och största sjukhus i Region Västerbotten tar vi emot många patienter med misstänkt ALS för diagnostik och bedömning. Vi bistår med genetisk rådgivning till både neurologiska kliniker och patienter, därtill utför vi genetisk testning för SOD1-mutationer hos ALS-patienter från hela världen. Vi är även ledande svenskt studiecenter för flertalet läkemedelsstudier.
Följande studier är pågående men tar inte längre emot nya patienter:
Flera nya studier är antingen på planeringsstadiet eller under förhandling med läkemedelsindustrin, Läkemedelsverket och Etikprövningsmyndigheten om att få tillstånd att utföra dem i Sverige. Vi återkommer med uppdaterad information om och när studierna har godkänts av Etikprövningsmyndigheten.
Förslag på kontaktpersoner: Peter Andersen och Lena Bylund
Anknuten personal: Peter Andersen, Karin Forsberg, Lena Bylund, Tina Björn, Elisabeth Müller Granberg, Nina Sundqvist och Ivar Winroth
Genom åren har vi samlat in mer än 14.000 olika prover från ALS-patienter och deras anhöriga. Med hjälp av dessa prover har vi i olika samarbeten upptäckt förändringar i flera gener som kan öka eller minska risken att utveckla ALS. Vårt fokus har under många år varit ALS som orsakas av förändringar i SOD1 genen. Genom analyser av våra insamlade prover har vi hittat ett tjugotal nya mutationer i SOD1-genen hos ALS-patienter, däribland D90A-mutationen som är den vanligast förekommande SOD1 mutationen i den skandinaviska populationen.
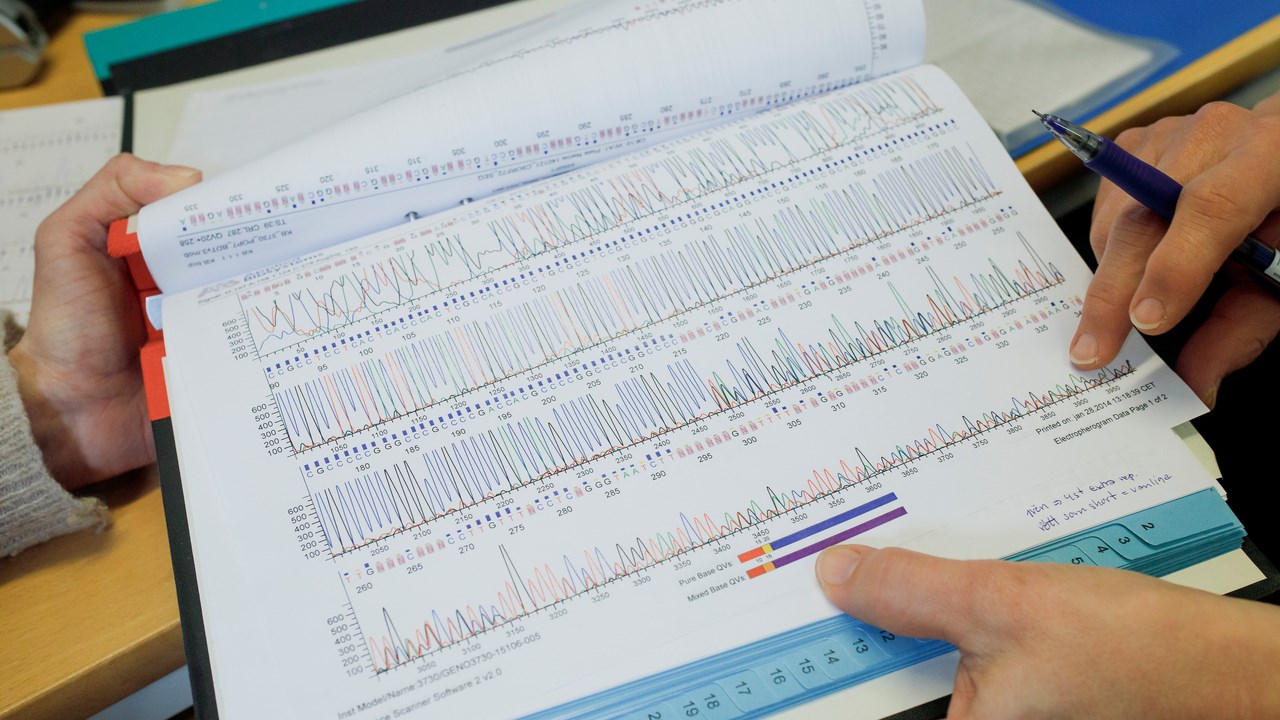
Sekvenseringsresultat från ett prov från en patient eller en forskningsperson. Resultatet kan visa om det finns en mutation känd för att orsaka ALS.
BildMattias PetterssonDessutom har vi varit med och utvecklat en kliniskt användbar metod för att identifiera mutationer i C9orf72-genen som är den idag vanligaste kända genetiska förändring som kan orsaka ALS. Vi utför rutinmässig screening för C9orf72-genen och SOD1-genen på alla ALS-patienter som behandlas av oss, skickar blodprov för analys via sin egen behandlande läkare, eller vill delta i vår forskning genom att lämna in forskningsprover. Vi är även deltagande part i det europeiska samarbetet Project MinE där hela arvsmassan från över 350 av våra forskningspersoner har sekvenserats i en så kallad hel-genom analys. Målet är att gemensamt studera och jämföra genetiska förändringar i tiotusentals ALS-patienter från Europa, Australien och USA för att kunna identifiera nya ledtrådar till vad som kan orsaka eller skydda mot ALS.
Läs mer om C9orf72- och SOD1-ALS
Förslag på kontaktpersoner: Peter Andersen, Angelica Nordin
Anknuten personal: Peter Andersen, Angelica Nordin, Helena Alstermark, Eva Jonsson, Soumyadeep Nandi, Emmy Sheltum Kockum, Ivar Winroth
Våra biokemiska studier innefattar cellens ämnesomsättning, proteiners funktion, och signalering mellan celler. Störst fokus läggs på hur olika proteiner som är involverade sjukdomsutveckling regleras, byggs upp och bryts ned, antar sin tredimensionella form och fungerar.
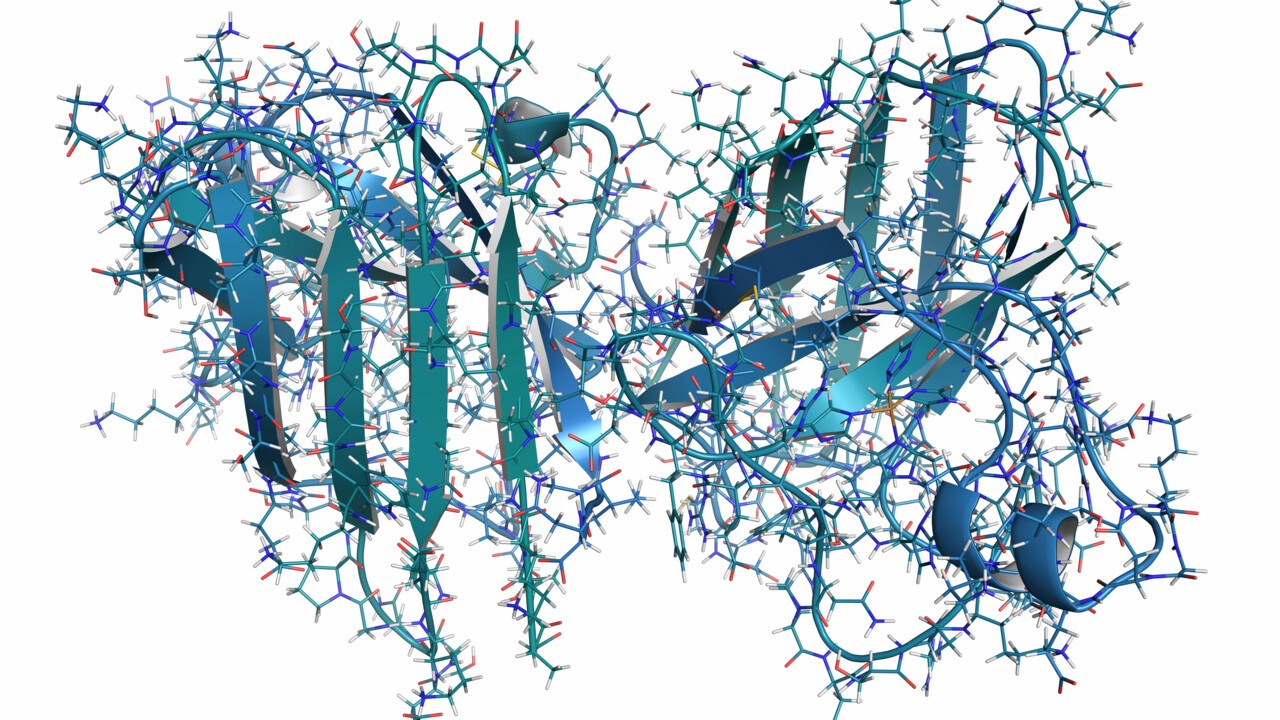
Illustration av SOD1 proteinets normala struktur
BildShutterstockSedan starten år 1993 har vi gjort noggranna studier av SOD1 proteinet och hur det orsakar ALS. Med åren har vi lärt oss att den enzymatiska, normala funktionen hos SOD1 ibland blir påverkad hos patienter med mutationer. Däremot verkar det inte främst vara detta som är orsaken till SOD1-relaterad ALS, eftersom även mutationer som inte förändrar proteinets förmåga att städa bort skadliga superoxider kan orsaka ALS. I stället verkar mutationerna försämra proteinets stabilitet. Resultatet blir ett protein som kan "tappa" sin tredimensionella form och anta nya, felaktiga former. Dessa nya former av så kallat "felveckat SOD1" får sjukdomsalstrande egenskaper och kan dessutom spridas mellan olika celler och tvinga andra SOD1-proteiner att anta samma sjukdomsframkallande form (sjukdomsassocierad struktur).
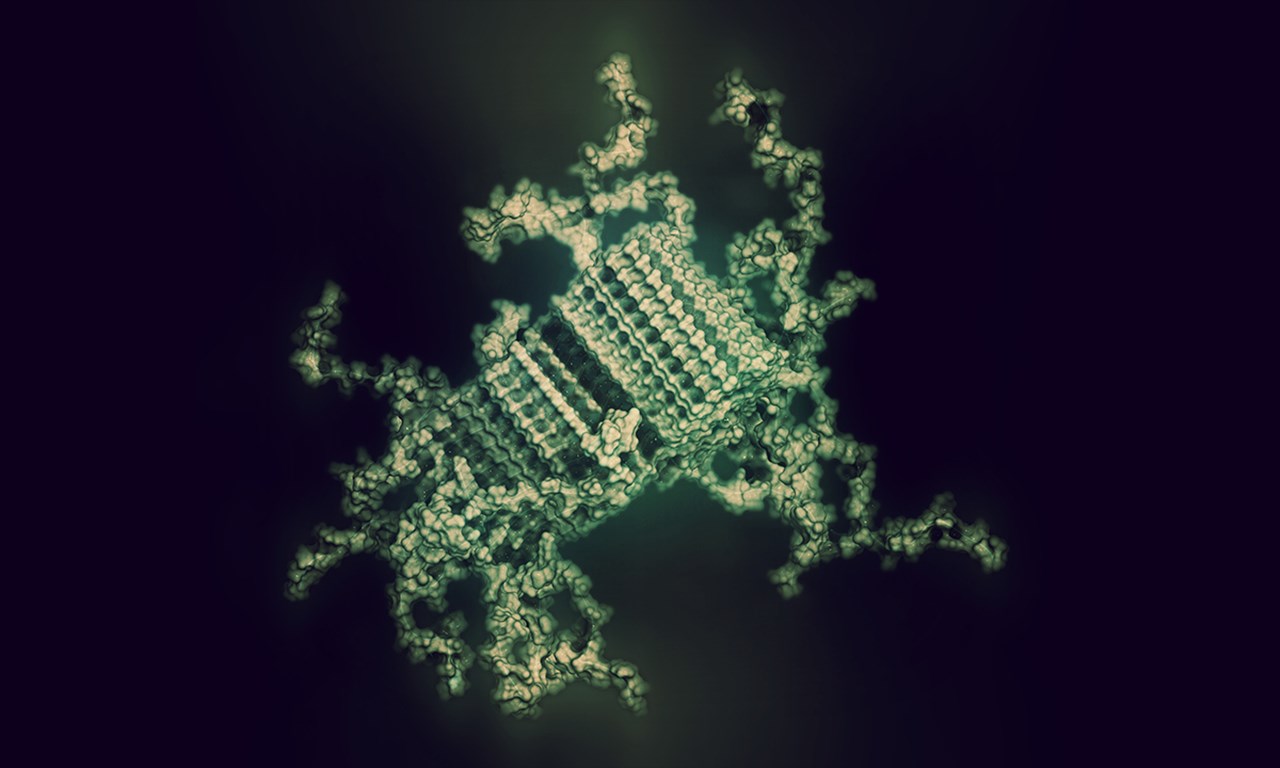
Illustration av hur felvecklade SOD1 proteiner kan se ut när de börjar forma de proteinfibriller som ansamlas och bildar de karakteristiska SOD1 klumpar man kan se i nervceller som skadas vid ALS. Vissa delar av proteinet är låsta i fibrillens kärna. Andra delar sticker ut och kan bindas av till exempel terapeutiska antikroppar.
BildShutterstock, Copyright (c) 2017 Shutterstock. No use without permission.I vårt biokemi-laboratorium mäter vi enzymaktivitet, analyserar felveckat SOD1 och försöker förstå de underliggande kemiska processerna som omvandlar proteinet från sin rätta, funktionella, form till den felveckade, sjukdomsalstrande formen.
Kontaktpersoner: Per Zetterström, Stefan Marklund
Anknuten personal: Helena Alstermark, Eva Jonsson, Agneta Öberg, Karin Hjertkvist, Ulrika Nordström, Caitin Henne, Laura Leykam
Biomarkörer kan sammanfattas som mätbara ämnen som proteiner, lipider eller andra molekyler, som till förändras eller läcker ut i blod eller ryggmärgsvätska vid sjukdom. Vi samlar in och undersöker blod och ryggmärgsvätska från patienter för att se hur olika molekyler och proteiner påverkas av sjukdomen.

Forskningsprover från patienter och anhöriga alikvoteras och kan användas för manga olika analyser. Det finns ofta stora individuella skillnader, vilket gör att vi behöver samla in prover från många patienter med samma typ av ALS för att kuna dra säkra slutsatser. Framför allt är det blodplasma, röda- och vita blodkroppar och cerebrospinal vätska som analyseras.
BildMattias PetterssonMålet är bland annat att hitta molekylära förändringar som kan fungera som kliniskt meningsfulla markörer som kan bidra till att enklare diagnosticera ALS, förutspå sjukdomsutveckling och visa om ett läkemedel bromsar sjukdomen.
Kontaktpersoner: Peter Andersen
Anknuten personal: Helena Alstermark, Eva Jonsson, Agneta Öberg, Karin Hjertkvist
Patologistudier innebär att man diagnosticerar och lär sig om sjukdomar genom analys av celler, vävnader och organ. Vi kan inte ta prover för att studera förändringar i de motoriska nervcellerna hos levande patienter utan att skada nervvävnaden. För att vi ska kunna studera de angripna cellerna i ryggmärgen och hjärnan är ALS-forskningen beroende av att patienter och deras anhöriga har haft kontakt med oss medan de varit i livet och uttryckt en önskan att få donera sin kropp till forskning efter döden.
Genom forskningsobduktionerna har vi dels möjlighet att i efterhand säkerställa att diagnosen ALS har varit korrekt. Dessutom kan vi lära oss mycket om sjukdomens påverkan på nervsystemet genom att studera hjärnan, ryggmärgen och musklerna hos avlidna patienter.

Paraffininbäddad vävnad snittas tunt för att läggas på glas inför infärgning med immunohistokemi.
BildMattias PetterssonEn viktig upptäckt som gjorts är att det finns ansamlingar av felaktiga eller fellokaliserade proteiner i de angripna cellerna i ryggmärgs-vävnaden. Kunskapen har bland annat lett fram till utveckling av behandlingsstrategier som riktas mot skadliga ansamlingar av felaktigt formade proteiner.
En av våra mest uppmärksammade upptäckter är att de flesta ALS-patienter, oavsett om de bär på en muterad SOD1-gen eller inte, har felveckat ihop-klumpat SOD1 i sina nervceller. Detta tyder på att SOD1 påverkar sjukdomsprocessen hos många fler ALS-patienter än de som bär på specifika SOD1-mutationer.
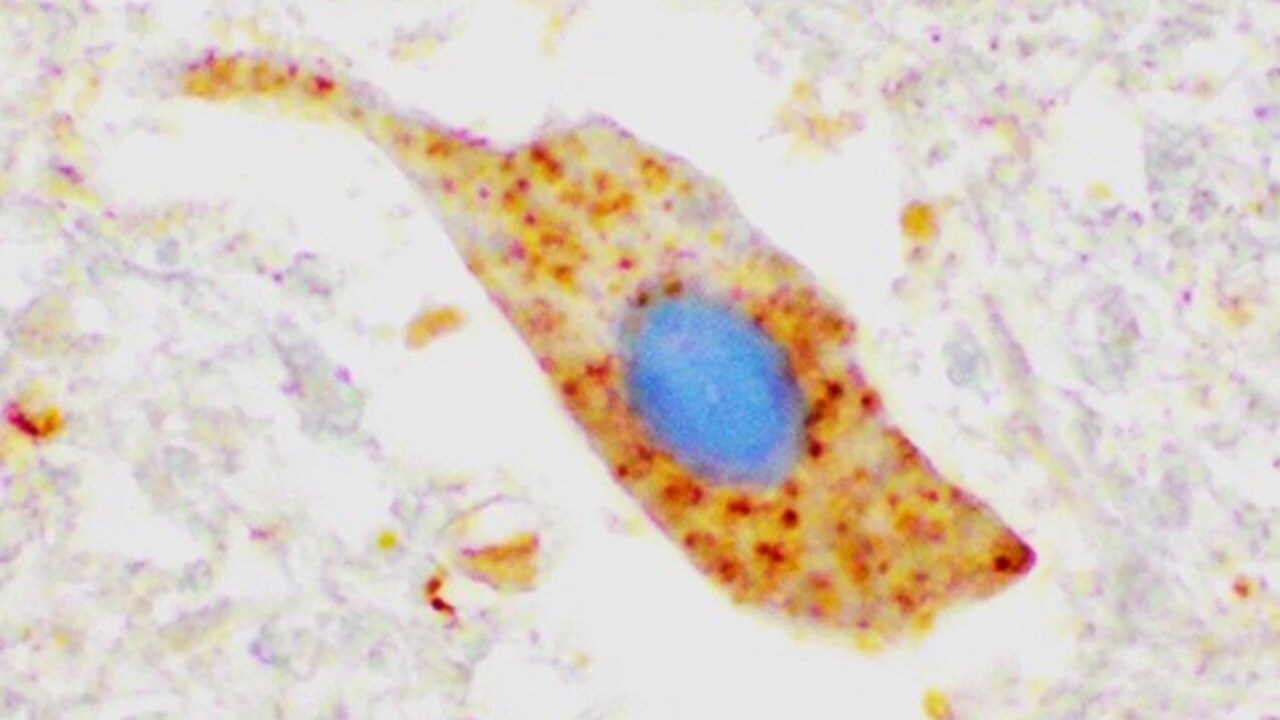
Bilden visar klumpar av SOD1 protein (färgat med brunt) i ett motorneuron i ryggmärgsvävnad från en ALS patient som inte har någon mutation i SOD1. Detta tyder på att proteinet kan vara inblandat i sjukdomsutveckling även vid sporadisk ALS.
BildKarin ForsbergKontaktpersoner: Thomas Brännström och Karin Forsberg
Anknuten personal: Ingrid Gustafsson, Emmelie Lidh, Matthew Marklund, Sara Rimpi, Erica Stenvall, Isabelle Sigfridsson
En svårbegriplig aspekt av ALS är att vissa muskler i kroppen påverkas mycket mindre av sjukdomen. Ögonmusklerna är ett sådant exempel. Även om vissa patienter efter lång respiratorvård kan få symtom på ögonförlamning så är flertalet patienter, oavsett om sjukdomen börjat i ansiktet eller i extremiteterna, relativt opåverkade i sina ögonrörelser.

De motoriska nervceller som styr ögats rörelseförmåga är särkilt motståndskraftiga vid ALS. Genom att studera dessa celler hoppas vi hitta ledtrådar om specifika egenskaper som kan förhindra eller bromsa sjukdomsutvecklingen.
BildMostphotos/Ruben JoyeÖgonmusklerna är högspecialiserade, evolutionärt bevarade muskler med egenskaper som inga andra muskler har. Om man bättre kan förstå ögonmusklernas bevarande vid ALS så kan man möjligen upptäcka nya sätt att behandla sjukdomen. Våra studier har visat att ögonmusklerna hos avlidna ALS-patienter är relativt välbevarade jämfört med andra muskler, men att det finns diskreta tecken på sjukdom. Sjukdomen verkar ge upphov till förändringar i signaleringen mellan ögonmuskler och deras nervceller och olika typer av ögonmuskelfibrer verkar drabbas olika hårt av sjukdomen, men än så länge går det inte att säga vilken/vilka av ögonmusklernas egenskaper som gör dem motståndskraftiga vid ALS.
Kontaktpersoner: Fatima Pedrosa Domellöf, Jingxia Liu
Eftersom vi inte kan studera förändringar i de motoriska nervcellerna i hjärnan och ryggmärgen hos levande patienter utan att skada nervvävnaden behöver vi modeller för att studera sjukdomsassocierade processer som kan orsaka ALS. Under åren har vi samlat hudbiopsier och odlat fram fibroblaster, dvs bindvävsceller eller stödjeceller från huden, från ALS patienter och deras anhöriga. Dessa celler kan hållas nedfrusna och sen tinas upp och odlas som cellkulturer i laboratorier.

Celler från hudbiopsier kan användas för att studera sjukdomsprocessen vid ALS utanför patientens egen kropp.
BildMattias PetterssonGenom cellförsök kan vi studera hur sjukdomen påverkar celler från ALS-patienter och hur cellerna och sjukdomsassocierade proteiner reagerar under olika förhållanden. Vi har kunnat studera hur patientens genetik och cellens miljö samverkar och påverkar sjukdomsfaktorer, som till exempel ansamlingen av felaktigt format SOD1. Vi har även omvandlat vissa av våra cellinjer till stamceller och fått dessa att mogna ut till motoriska nervceller. Detta gör att vi kan studera sjukdomsprocessen i celler utanför patientens egen kropp. Cellförsöken har gett oss inblick i vilka faktorer som bidrar till ökad felveckling, men även vilka system inuti cellen som är viktiga för att bryta ner felvecklat SOD1 protein.
Kontaktpersoner: Peter Andersen, Ulrika Nordström
Anknuten personal: Eva Jonsson, Isil Keskin
För att bättre förstå sjukdomsutvecklingen vid ALS bedriver vi djurförsök inom ramen för Umeå Centre for Comparative Biology (UCCB). Vi har framställt flera genetiskt modifierade musstammar som uttrycker humant SOD1 med olika mutationer som kan orsaka ALS hos människor. Uttryck av muterat SOD1 protein resulterar i en ALS-liknande sjukdom hos mössen som är mycket lik den som drabbar människor.
Tillgången till flera olika musstammar låter oss studera olika stadier av sjukdomsutvecklingen i nervsystemet på ett sätt som vore omöjligt hos patienter. Olikheterna mellan musstammarna speglar också en motsvarande olikhet mellan olika patientgrupper, vilket hjälper oss att undersöka varför sjukdomen ibland har ett aggressivt förlopp och ibland ett mer långsamt.
Vi har även genom injektionsexperiment skapat en musmodell för att studera spridning av ihopklumpat felvecklat SOD1. Modellen ger oss möjlighet att studera effekt och spridning av proteinansamlingar och deras roll i sjukdomsutvecklingen.
Alla djurförsök har godkänts av djurförsöksnämnden och sker i enlighet med svenska lagar och EU-bestämmelser om försöksdjursverksamhet.



